
| PDB Code and Chain ID |
Total num. residues |
Interface num. residues |
ProBiS detected united binding site | Sensitivites for different binding sites types | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P-value | Num. residues |
SP | SE | Small- ligand |
Protein- protein |
DNA | Other | |||
| 1all.A | 160 | 68 | 2.20E-06 | 48 | 0.708 | 0.500 | 0.483 | 0.523 | - | - |
| 1apm.E | 338 | 57 | 2.50E-07 | 101 | 0.337 | 0.596 | 0.774 | 0.525 | - | - |
| 1aze.A | 56 | 23 | 7.60E-02 | 15 | 0.600 | 0.391 | - | 0.250 | - | 0.714 |
| 1bnc.A | 433 | 75 | 1.60E-06 | 123 | 0.317 | 0.520 | 0.788 | 0.310 | - | - |
| 1daa.A | 277 | 82 | 2.10E-02 | 70 | 0.400 | 0.341 | 0.885 | 0.143 | - | - |
| 1dfj.E | 124 | 46 | 4.60E-02 | 36 | 0.500 | 0.391 | 1.000 | 0.378 | - | - |
| 1dfj.I | 456 | 49 | 1.60E-01 | 117 | 0.137 | 0.327 | - | 0.327 | - | - |
| 1efu.A | 364 | 92 | 5.50E-14 | 112 | 0.518 | 0.630 | 1.000 | 0.590 | - | - |
| 1g3n.A | 293 | 89 | 7.00E-01 | 87 | 0.287 | 0.281 | 0.594 | 0.154 | - | - |
| 1g3n.B | 155 | 28 | 1.00E+00 | 46 | 0.043 | 0.071 | - | 0.071 | - | - |
| 1g3n.C | 233 | 38 | 6.60E-03 | 62 | 0.274 | 0.447 | - | 0.447 | - | - |
| 1got.A | 326 | 63 | 2.90E-05 | 98 | 0.337 | 0.524 | 1.000 | 0.167 | - | - |
| 1got.B | 339 | 123 | 9.90E-01 | 93 | 0.269 | 0.203 | - | 0.203 | - | - |
| 1hcg.A | 229 | 59 | 1.10E-01 | 69 | 0.319 | 0.373 | 0.615 | 0.182 | - | - |
| 1k9o.E | 223 | 37 | 9.20E-04 | 68 | 0.294 | 0.541 | - | 0.541 | - | - |
| 1k9o.I | 376 | 94 | 1.30E-05 | 98 | 0.418 | 0.436 | - | 0.436 | - | - |
| 1lw6.I | 63 | 18 | 5.50E-01 | 13 | 0.308 | 0.222 | - | 0.222 | - | - |
| 1rrp.A | 204 | 100 | 4.30E-01 | 61 | 0.508 | 0.310 | 0.848 | 0.083 | - | - |
| 1tco.B | 169 | 108 | 1.90E-05 | 45 | 0.889 | 0.370 | 0.791 | 0.129 | - | - |
| 1ugh.E | 223 | 35 | 8.60E-04 | 41 | 0.341 | 0.400 | - | 0.400 | - | - |
| 1ytf.A | 180 | 59 | 2.20E-08 | 51 | 0.647 | 0.559 | - | 0.500 | 0.630 | - |
| 1ak4.A | 165 | 22 | 1.80E-07 | 45 | 0.38 | 0.77 | - | 0.77 | - | - |
| 1ay7.A | 96 | 21 | 4.00E-06 | 24 | 0.58 | 0.67 | - | 0.67 | - | - |
| 1bvn.P | 496 | 60 | 2.00E-07 | 141 | 0.25 | 0.58 | 0.83 | 0.5 | - | 1 |
| 1efn.B | 104 | 25 | 1.30E-05 | 29 | 0.55 | 0.64 | - | 0.64 | - | - |
| 1fqj.B | 133 | 31 | 2.50E-04 | 37 | 0.46 | 0.55 | - | 0.55 | - | - |
| 1gcq.C | 69 | 30 | 9.60E-01 | 20 | 0.3 | 0.2 | - | 0.2 | - | - |
| 1gpw.A | 253 | 68 | 4.50E-03 | 71 | 0.39 | 0.41 | 0.39 | 0.43 | - | - |
| 1gpw.B | 200 | 28 | 8.60E-02 | 60 | 0.2 | 0.43 | - | 0.43 | - | - |
| 1grn.A | 191 | 48 | 2.60E-09 | 56 | 0.55 | 0.65 | 0.85 | 0.48 | - | - |
| 1grn.B | 197 | 44 | 6.30E-03 | 57 | 0.35 | 0.46 | - | 0.46 | - | - |
| 1h1v.A | 368 | 124 | 1.90E-01 | 98 | 0.38 | 0.3 | 0.82 | 0.08 | - | - |
| 1h1v.G | 327 | 67 | 2.00E-05 | 95 | 0.36 | 0.51 | 0.54 | 0.47 | - | - |
| 1ktz.A | 82 | 31 | 8.10E-02 | 28 | 0.5 | 0.45 | - | 0.45 | - | - |
| 1pxv.A | 183 | 40 | 1.20E-03 | 53 | 0.38 | 0.5 | - | 0.5 | - | - |
| 1pxv.C | 111 | 31 | 1.40E-03 | 32 | 0.5 | 0.52 | - | 0.52 | - | - |
| 1r8s.E | 187 | 47 | 4.60E-02 | 48 | 0.35 | 0.36 | 0.64 | 0.36 | - | - |
| 1s1q.A | 137 | 22 | 1.00E+00 | 36 | 0 | 0 | - | 0 | - | - |
| 1udi.E | 227 | 34 | 1.20E-05 | 65 | 0.32 | 0.62 | - | 0.62 | - | - |
Heterotrimeric G proteins relay hormonal signals from transmembrane
receptors to intracellular effectors. They consist of three subunits:
alpha, beta, and gamma (see Fig. 1). When a hormone reacts with its
extracellular receptor, the intracellular membrane-bound G protein in
turn binds to this hormone-receptor complex. The GDP which is bound to
the G protein at rest is replaced by the GTP &bull Mg2+
complex, while the beta-gamma subunit splits off. The free alpha
subunit then activates (or inhibits) various secondary effectors.
The alpha subunit itself has two domains. One adopts a transducin insertion domain fold and the other a P-loop containing nucleoside triphosphate hydrolase fold (SCOP classification of protein structures) (see Fig. 1). P-loop or a phosphate-binding loop is an ATP or GTP- binding site motif found in many nucleotide-binding proteins. It is a glycine-rich loop preceded by a beta sheet and followed by an alpha helix. It interacts with the nucleotide phosphate groups and with the Mg2+ ion that coordinates the β- and γ-phosphates in GTP. Upon nucleotide hydrolysis the P-loop does not significantly change conformation, but stays bound to the remaining phosphate groups. Conversely, the alpha-beta binding site region does change its conformation significantly in this process (encircled in Fig. 1).
In our study, we used the crystal structure of the alpha subunit (PDB
ID: 1got, Chain Id: a) bound to the beta-gamma subunit and to a GDP
molecule as the input structure for the ProBiS algorithm. Due to
prominent flexibility and the presence of several binding sites this
structure is ideal for testing ProBiS's ability to detect binding
sites. It was compared to nearly 23,000 protein structures in the
current non-redundant protein database (nr-PDB), from which ProBiS
retrieved 408 locally similar protein structures. Binding sites
predictions were obtained by calculating the structural conservation
scores for the query protein residues based on these retrieved
structures.
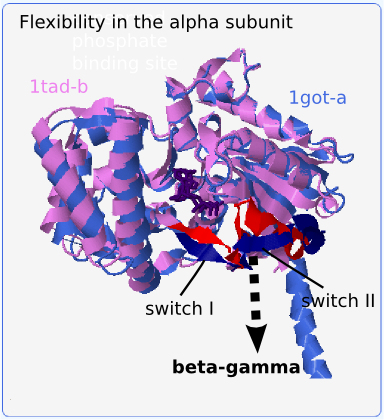
We can observe the conformational changes in the protein-protein binding site if we superimpose the bound structure (1got-a) onto the homologous (>90% sequence identity) but unbound alpha subunit (1tad-b) (see Fig. 2; the flexible segments are red and dark blue). The loop/alpha helix residues Val196-Glu212 (referred to as switch II) adopt very different conformations in these two structures. Upon binding, the alpha helix partially unfolds between Arg201 and Arg204 and the whole segment tilts up to 8 Å away from the protein core and towards the beta-gamma subunit. This move is accompanied by the shift of nearby residues Val175-Ile180 (referred to as switch I) by around 5 Å. Since structures of alpha subunits in PDB may be bound or unbound, it is necessary to be able to find similarities in flexible segments when comparing these structures.
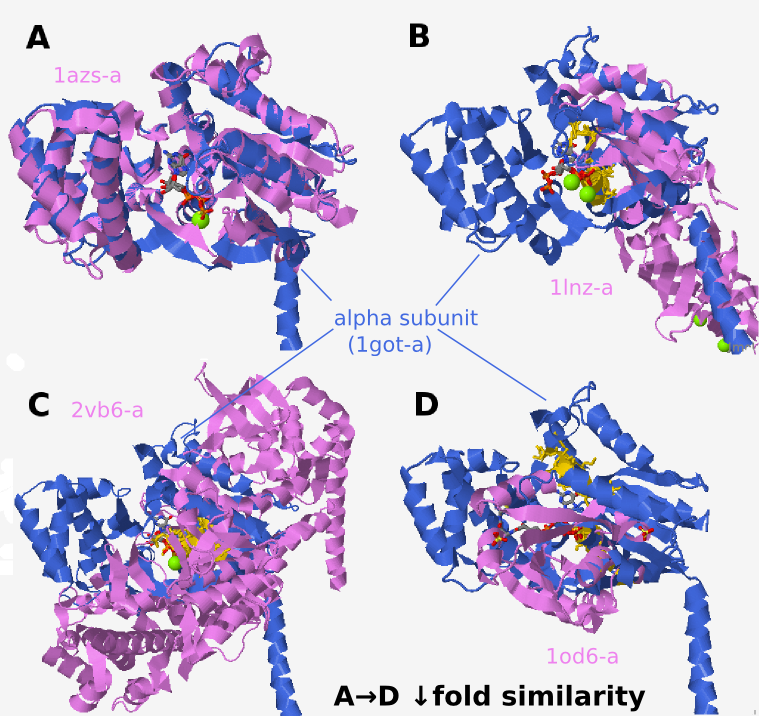
Retrieved structures resemble the query protein to different
extents: some resemble both alpha subunit domains (Fig. 3A),
some resemble only one (Fig. 3B), while others share no fold
similarities to either of the two domains, except for similar
binding residues (Fig. 3C and D). The structurally aligned
residues which are usually present in the GDP binding region are
colored gold in Fig. 3. Although the two protein backbones could
at times be aligned, e.g. Fig. 3B, the strongest structural
similarity is usually restricted to binding site residues. This
is the main difference between local and global alignment of
proteins; the latter seek similarity in protein backbones. In
contrast, ProBiS aligns proteins on the most similar local
surface interaction patterns. Not even small segments of
backbone need to be similar. ProBiS can make use of these
locally similar motifs to detect structurally conserved binding
sites. Even though in minority, the detected cross-fold
structural similarities (Fig. 3C and D) are important
contributors to the final conservation scores and present
interesting cases for further discussion.
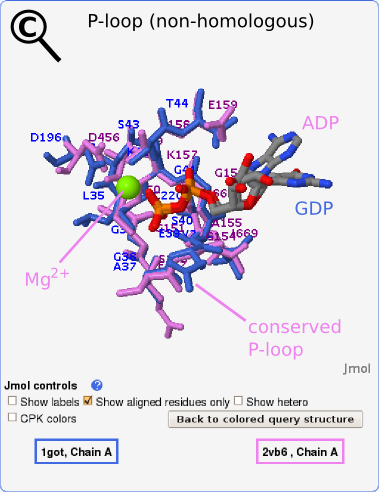
Exploring structures with decreasing fold similarities to the query we find structures which share only local similarities with our query protein. These similarities are detected especially in binding sites as in the case presented in Fig. 3C. A detailed view of the aligned binding site region is shown in Fig. 4. Here, a similarity between the GDP binding site in the alpha subunit (query) and the ADP binding site of myosin VI (2vb6-a) was detected. The two compared proteins are non-homologous (sequence alignment with BLAST yields E-value of 0.74 and global structural alignment with MAMMOTH produces no meaningful alignment), yet both possess a conserved P-loop which is responsible for the binding of nucleotide phosphate groups. The phosphates are superimposed by the transformation and adopt exactly the same conformation in both structures, as shown in Fig. 4. The Mg2+ ion in the aligned structure is also shown.
Perhaps the most interesting are structures which are not fold similar
to the query, but have through the evolution converged to a similar
binding site. However, false positives which are due to chance
similarities in non related proteins may be mistaken for a real
binding site similarity in this case. It is difficult to distinguish
real from artifact similarities if no ligands are present in the
structure or the binding site location is not known. However, the
alignment scores (especially E-value), which ProBiS assigns to each
local alignment, try to foresee false positives. In one of the
retrieved structures (1od6-a, Fig. 3D) we could confirm that
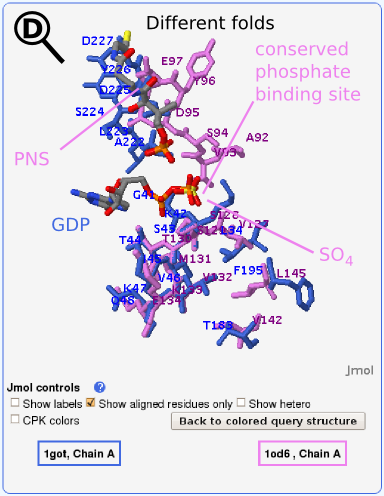
a similar ligand (ADP) binds to it as in our query
(GDP). Phosphopantetheine adenylyltransferase, which by the SCOP
classification adopts an adenine nucleotide alpha hydrolase-like fold
(violet in Figs. 3 and 5) is aligned to the query structure, G protein
alpha subunit, which by SCOP classification adopts a P-loop containing
nucleoside triphosphate hydrolase fold (blue in Figs. 2 and 4). The
E-value for this alignment is 1.1 ⋅ 10-7. The detailed view of the
aligned binding site region is shown in Fig. 5, where ADP is not shown
because it was not present in the crystal structure. The similarity
between binding sites is due to a similar function of the two compared
proteins, i.e., phosphate binding. Our attention was first attracted
by the
SO42-
ion, which aligns almost perfectly with the the terminal
phosphate group in GDP from the query structure (see
Fig. 5). Subsequent comparison with related PDB entries (e.g.,
1gn8-a), revealed that the
SO42-
position is usually occupied by an
ADP phosphate group. The alignment includes residues in the P-loop and
alpha helix preceding the loop in the alpha subunit (query structure)
with the alpha helix in the similar structure.
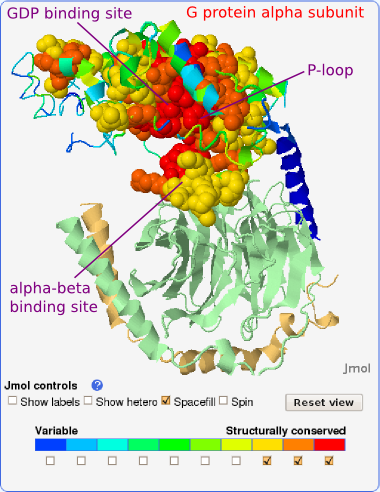
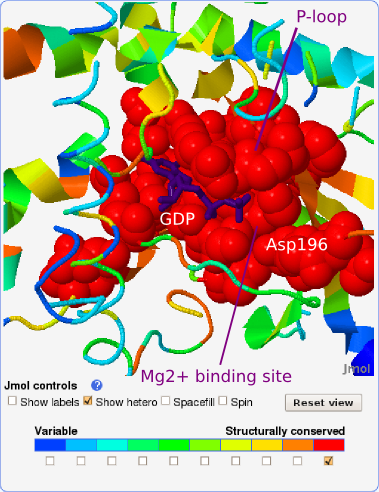
We consider all residues with local structural conservation scores of
7 or more as the prediction of binding sites. Using this definition,
the GDP (and Mg2+) binding site on the alpha subunit
was predicted
with the sensitivity of 0.852. In other words, 85.2% of the binding
site was found to be structurally conserved (see Fig. 6 and zoom-in on
the most conserved GDP binding site in Fig. 7).
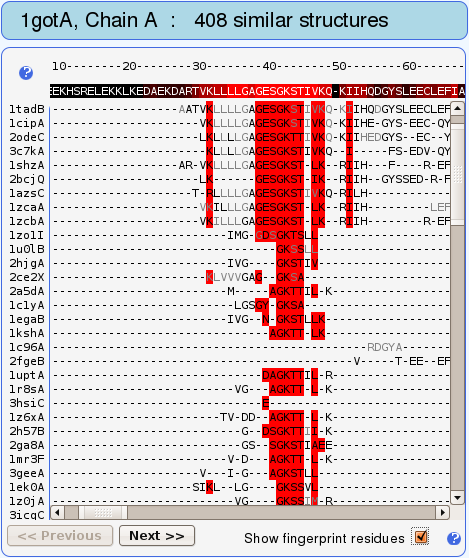
Not surprisingly, the P-loop with sequence GAGESGKS (residues
Gly36-Ser43) is designated as the most structurally conserved
part of this protein (see Fig. 7). These so called fingerprint
residues have been found to be conserved in the best structural
alignments (with highest alignment lengths to the query) and are
used to scan the remaining alignments for similar
motifs. Structures having the same fingerprint motif are
thereby retrieved.
Fingerprints are vertical stripes of residues highlighted with
red color in the structure-based sequence alignment (see Figs. 8
and 9).
Another fingerprint residue, conserved in
most of the retrieved proteins, is Asp196 (see also Fig. 9). The
structurally conserved Asp196 is part of a conserved sequence
motif, DXXG, found in all regulatory GTP binding proteins, and
forms the amino-terminal segment of switch II. Aspartate 196
serves to connect the Mg2+ site to switch II via a
water molecule that is bound to the Mg2+ ion.
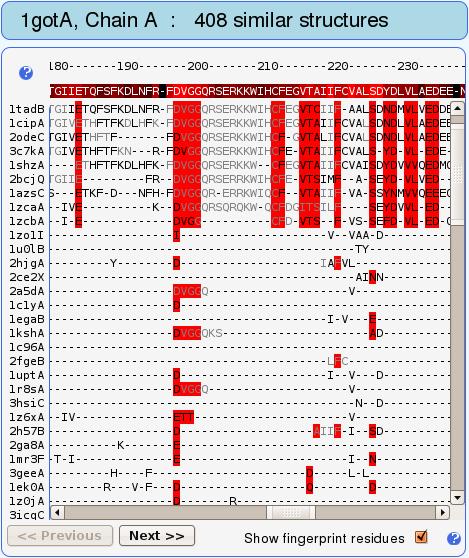
The protein-protein binding site on the alpha subunit, i.e., the alpha-beta binding site which spans mainly the switch II region, was predicted with the sensitivity of 0.389. In other words, 38.9% of this binding site is structurally conserved (see Fig. 6). This binding site has a dual role: it binds the beta-gamma subunit as is the case in our query crystal structure, but it also interacts with adenylate cyclase after the beta-gamma subunit has split off (e.g., 1cul). Our observations concern only the alpha-(beta-gamma) binding. The considerably lower structural conservation compared with conservation in GDP binding site could be a consequence of the C-terminal alpha helix (blue on Fig. 5), which is also a part of alpha-beta binding site, being missing in most of the retrieved structures. On the other hand, in this study protein-protein binding sites have generally been found to be less well conserved than binding sites for small ligands. Still, residues Val197-Gln200 in the flexible part of the binding site are recognized as well conserved (structural conservation score: 9) and residues Arg201-Glu212 as moderately conserved (structural conservation score: 7-8) (see Figs. 5 and 9). To distinguish the conserved residues, which adopt different conformations (i.e., lie on flexible backbone segments) from those that adopt the same conformation (i.e., can be rigidly superimposed) in compared structures, they are colored gray and black in the structure-based sequence alignment, respectively (see Figs. 8 and 9).